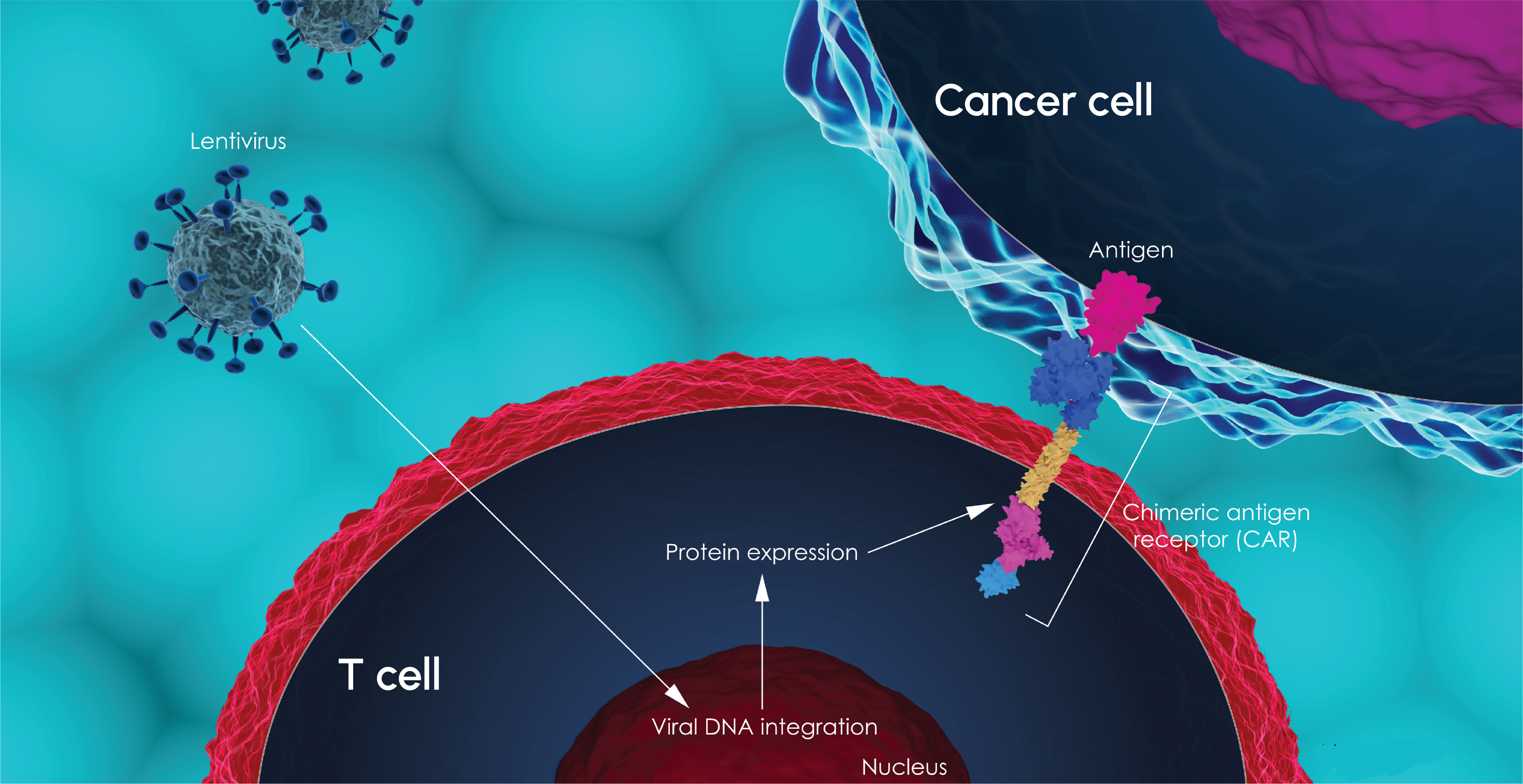
-1.png)
At Advanced Therapies Week 2026, our Chief Technology Officer, Gregg Nyberg, joined a panel of industry leaders to discuss how advanced therapy developers are navigating the path from early-stage process development to commercial manufacturing. The conversation reinforced something we see in nearly every program we support: the hardest part of scaling a cell or gene therapy isn't any single inflection point. It's the series of small, often quiet decisions made years earlier that determine whether the next stage is a smooth translation or a costly reinvention.
The Panel
|
VP, Technical Operations Caribou Biosciences |
Lavakumar (Kumar) Karyampudi, Ph.D. Senior Director, Cell Therapies Moffitt Cancer Center |
VP, Marketing & Experience, Life Sciences Emerson |
|
Chief Technology Officer Landmark Bio |
SVP & Head of Technical Operations Tolerance Bio |
This post distills what we believe are the most important of those decisions, drawing on the panel discussion, our own experience working across cell therapy, gene therapy, and complex biologics programs, and the patterns we see repeated in programs that land successfully in the clinic and beyond.
Tech Transfer Planning Starts on Day One
For most small biotechs, internal GMP manufacturing isn't realistic. Some form of technology transfer to a contract partner is effectively inevitable. That reality has a design implication most programs underestimate: your process and analytical development work should be built with a receiving site in mind from the earliest bench experiments.
This doesn't mean every Phase 1 process needs to be commercial-grade. It means every Phase 1 process needs to be GMP-compatible and legible to a partner who wasn't in the room when the decisions were made. The programs that struggle most at tech transfer aren't the ones with unusually complex biology. They're the ones where knowledge was captured informally, assays were optimized for a specific analyst, and critical process decisions were never written down in a form that survives a handoff.
Advanced therapies are also inherently multi-transfer products. A process developed in an academic setting or at a translational CDMO will almost certainly move again, to scale, to a new site, or to a commercial partner. Every early decision is a deposit into that future. A fellow panelist framed it as starting with the end in mind, and that framing is exactly right: the question is never whether your process will be transferred, only how many times.
What this looks like in practice
-
Process development documentation that reads as clearly to an external team as it does to the scientists who ran the work.
-
Analytical methods that aren't dependent on a single operator or a single instrument configuration.
-
A critical quality attribute framework that's reasonable for the program's stage but robust enough that you can trust your clinical signal when it arrives.
-
Explicit decisions about what's locked down for Phase 1 and what remains flexible, documented so the next team inherits the reasoning, not just the outcome.
Build vs. Buy: The Math Has Changed
Five or six years ago, in the boom years for biotech, many advanced therapy developers built their own manufacturing facilities. Capacity was constrained, capital was plentiful, and owning the clean room felt like the safer bet. The landscape today looks different. Capacity is widely available. Capital is more constrained. And the dose volumes required for early clinical work rarely justify the cost of operating a dedicated facility that sits partially empty between campaigns.
For the vast majority of early-stage programs, outsourcing to the right partner is now the right economic answer. But we'd argue it's also the right strategic answer, and that second point gets less attention than it deserves. If an independent manufacturing partner can reproduce your process successfully, the value of your program rises. "Only we can make this" isn't a strength story for investors, for potential acquirers, or for the long-term viability of a therapy. Reproducibility at arm's length is.
"If you over-invest early, you may not have a company to get to the next stage. But you also don't want to cut corners and incur technology debt that forces expensive rework later. Quality-by-design helps you focus investment on the attributes most likely to matter."
Gregg Nyberg, Chief Technology Officer, Landmark Bio
Choosing the right partner for your stage
The criteria that matter most shift as your program matures. For first-in-human work, phase-appropriate capabilities, scientific engagement, and the right level of attention often matter more than massive commercial infrastructure. As programs move toward pivotal and commercial, the filters shift: scalable GMP infrastructure, modality-specific experience, regulatory track record, and a partnership philosophy that treats tech transfer as a collaboration rather than a one-way handoff.
What we've seen work particularly well is engaging with a manufacturing partner early, even before the official tech transfer kickoff. A team supply run or an early engineering campaign tells you more about how a process will behave in GMP than any amount of documentation review can. It also surfaces the questions that are better asked now than at BLA stage.
Phase Appropriateness Is a Resource-Allocation Decision
One of the hardest judgment calls in advanced therapy development is deciding what to invest in and when. Programs that over-specify their processes at Phase 1 often arrive at the clinic years later than programs that prioritized getting meaningful human data first. Programs that under-invest in their CQA framework can't trust the signal they see when the data does arrive.
We don't believe there's a universal answer to this trade-off. What we believe is that programs need to make it deliberately, not by default. A fully qualified potency assay, for example, is not a regulatory requirement for a Phase 1 filing. It's critical for mechanism-of-action confidence and for commercial readiness, but the panel raised a fair point: programs that chase perfect assays before first-in-human sometimes fall behind peers who accepted a simpler early assay and planned for comparability work later. The right answer depends on the biology, the modality, and the program's funding runway.
One framing from the panel that resonated with us: cost, quality, and timeline are three levers you can only optimize two of at once. Want speed and quality? You'll pay for it. Want low cost and speed? Quality will suffer. Want cost and quality? Your timeline stretches. Every program is continuously tuning among those three, and the right balance shifts as the program matures.
Quality-by-design as a focus tool
Quality-by-design is sometimes treated as a commercial-stage activity, but it's most valuable earlier, when resources are tightest. Used well, it's not about maximizing quality everywhere; it's about identifying which quality investments have the highest leverage and concentrating effort there. Not every step in a process carries equal weight. Not every attribute is equally likely to be critical. The sooner a team can make that distinction, the more efficiently it allocates both time and money.
The Hardest Thing to Transfer Is Tacit Knowledge
Batch records and SOPs are necessary, but they're the easy part. The knowledge that actually determines whether a receiving site succeeds is tacit: the judgment calls, the "how we handle this edge case" intuitions, the institutional memory that accumulates over years of running similar processes. That kind of knowledge rarely lives in documents. It lives in people.
The programs that transfer well are the ones that treat tacit knowledge as something to be deliberately moved, not something that will transfer on its own. In practice, that means three things.
- Document what's usually undocumented. What has worked on similar programs. What hasn't. What your site has learned that isn't in the batch record.
- Invest in shadowing, not slide decks. Receiving-site staff spend time at the sending site. Sending-site staff are present on the floor for the first validation runs. The camaraderie that builds between teams is not a nice-to-have; it's what carries the partnership through the first deviation.
- Build redundancy into expertise. A single long-tenured expert is a vulnerability, not an asset. Institutional knowledge has to live across multiple people to survive staff changes, and those changes happen more often than anyone plans for.
One panelist put it sharply: you can't PowerPoint your way through a tech transfer. That matches what we see. The transfers that feel easy in the planning phase but fall apart in execution are almost always the ones that leaned on presentations and documents rather than people spending real time together.
Digital Infrastructure: Start Simple, Start Early
Digital transformation in manufacturing is often framed as a commercial-stage problem. We'd argue the opposite. The bridge to commercial manufacturing doesn't start at the end of the bridge; it starts at the bench. The companies that struggle most with digital integration late aren't the ones that picked the wrong platform. They're the ones that never developed the habit of capturing decisions alongside data in the first place.
You don't need an enterprise data fabric at the research stage. What you need is a discipline around recording why decisions were made, not just what the outcome was. Why did the team change buffer composition? Why did they settle on a particular cell density? That reasoning is what makes future scale-up a translation problem rather than a re-invention problem.
Structured spreadsheets are a legitimate starting point. The sophistication grows with the program. But the habit has to start early, because by the time you need the data for a comparability package or a regulatory filing, there's no way to reconstruct what wasn't captured.
Commercial Readiness Is Really About Regulatory Strategy
A detail that often surprises developers new to advanced therapies: every mid-program change carries a comparability cost. And at commercial stage, that cost can be substantial enough to force changes in approach, or in extreme cases, to force additional clinical work.
The programs that navigate this well are the ones that treat regulatory strategy as a Phase 1 concern, not a BLA concern. A program that plans for minimal process changes between Phase 1 and pivotal can sometimes carry early clinical data all the way through filing. A program that changes its process every time something improves may find that earlier data no longer applies.
This isn't an argument for never changing a process. Processes improve, and sometimes those improvements are essential. It's an argument for making those changes deliberately, with a clear view of what they cost in comparability work and how that cost compounds across a program's life cycle.
Tech Transfer Is Owned Jointly, by Design
We're sometimes asked who owns the success of a tech transfer: the sending site or the receiving site? The honest answer, and the answer we believe in, is that neither owns it alone. That's the point.
What works is explicit joint ownership, with three things established from day one: governance, accountability across each sub-function (manufacturing, analytics, quality), and a defined path of escalation. Without clarity on any of those, the transfer drifts. Issues become blame. The relationship frays at exactly the moment both sides most need it to hold.
The partnerships that work best are the ones where both sides know what success looks like, what their specific piece of it is, and how to raise problems early enough that they can be solved rather than managed.
What the FDA's Recent CMC Flexibility Means in Practice

The FDA's recent announcement signaling greater flexibility on CMC for cell and gene therapy was welcome news to the panel, and to us. The question, as always, is how it plays out in practice. The announcement is a statement of intent, not a guidance document, and much of what's in it represents a commitment to use tools that already exist, quality-by-design approaches, prior knowledge, platform data, more consistently.
Our read: this is a genuinely constructive signal, particularly for programs approaching BLA. But programs should not plan as if the regulatory bar has shifted. Plan as if the tools for engaging flexibly with the agency have been reaffirmed, and prepare to use them well. The strongest CMC packages still come from programs that did the underlying process and analytical work carefully, not from programs that counted on flexibility to compensate for gaps.
How We Think About This
The thread that runs through all of this, from the earliest process development decisions to the partnerships that carry a program to commercial, is that scaling an advanced therapy is never a single event. It's a long sequence of decisions, most of which are made quietly, years before their consequences become visible.
At Landmark Bio, we think of our role as helping developers make those decisions deliberately, with the benefit of what we've learned across the programs we've supported. The work we care most about isn't the headline tech transfer moment. It's the hundred small decisions in the year before it that determine whether the transfer feels like a translation or a reinvention.
If your program is approaching one of those decisions, we'd welcome a conversation.
Full Session Transcript
Read the full panel transcript
Panelist Introductions
Justin Skoble, Caribou Biosciences: Hi, I'm Justin Skoble, the Vice President of Technical Operations at Caribou Biosciences. We're developing CRISPR genome-edited allogeneic CAR-T cells. I oversee our process development, analytical development, and manufacturing teams and our supply chain function at Caribou. We're really trying to expand access to CAR-T cells by making them off the shelf.
Lavakumar (Kumar) Karyampudi, Moffitt Cancer Center: Hello, everyone. I'm Kumar Karyampudi. I'm a Senior Director of Cell Therapies at Moffitt Cancer Center. I oversee a cGMP cell and vector manufacturing group at Moffitt. It's a group of 80 people that handles a wide variety of cell therapy products as well as early-stage vector manufacturing. I'm glad to be part of this panel and looking forward to an exciting discussion.
Nikki Bishop, Emerson: Good morning, everyone. Nikki Bishop. I'm the Vice President of Marketing and Customer Experience for Emerson's life sciences software portfolio. All this week when I meet somebody, they say, "Emerson, oh, that Emerson." So I'll go ahead and say, yes, that Emerson. Emerson Electric is the larger company, but we focus on manufacturing and automation software solutions for optimization in manufacturing.
Gregg Nyberg, Landmark Bio: Good morning. I'm Gregg Nyberg, Chief Technology Officer at Landmark Bio. Landmark is a U.S.-based contract development and manufacturing organization that specializes in the advanced therapy space, especially in the translational area. We were recently acquired by Artist Biosolutions, so we're part of the Artist Biosolutions group.
Yeh-Chuin Poh, Tolerance Bio: Hi, everyone. I'm Yeh-Chuin Poh. Tolerance Bio is a new startup, only been around for a year and a half to two years. We're a therapeutic company with two different platforms. The first is a regenerative medicine platform using iPSCs differentiated into thymic cells to regenerate the thymus. The other is a monoclonal antibody platform, using a monoclonal antibody to preserve thymic involution and prevent atrophy.
Part 1: Development Through Phase 1
Moderator: Let's start with tech transfer. How soon do you start planning a tech transfer? Is it something that's in mind from the beginning, or do you kick it off when the need arises?
Justin Skoble: Planning for tech transfer is something you have to do if you want to actually develop a drug. Most small biotechs and startups can't afford their own manufacturing, so you'll have to transfer your process to a CDMO. It's really important to invest early in process development and analytical development internally, so that you know how to make your product, how to analyze it, what "good" looks like, and how to maintain critical quality attributes. It doesn't have to be perfect for Phase 1, but it needs to be at least somewhat compatible with GMP principles. Further tech transfers happen later as you scale up.
Yeh-Chuin Poh: I've developed several cell therapy products over my career, and tech transfer is always something people talk about. Some companies use stage-gating internally; when you hit certain milestones, you start the transfer process. But there's a balance. You can't wait for the researchers to finish before starting tech transfer. You need to start with the end in mind and develop something that's manufacturable. That said, if you try to develop something for commercial applications at the outset, the cost will be extreme and most startups can't absorb it. You need to be phase-appropriate: build something translatable to commercial, but without spending all your resources up front before you have clinical data.
Kumar Karyampudi: Just to add, we all know for cell therapy products, there are always multiple tech transfers inherent to any product. From day one of process development, you have to keep in mind that this product will be transferred somewhere else to scale up or support late-stage manufacturing.
Moderator: How do you decide between outsourcing the work and manufacturing in-house? And if you do outsource, how do you select the right CDMO?
Yeh-Chuin Poh: This is a big question, build versus buy. Five or six years ago, when biotech was booming, you saw many companies building their own manufacturing facilities because there was a capacity constraint. Today, many of those facilities are sitting empty. There's excess capacity, and you didn't need to spend all the capital up front. A lot of CDMOs are still operating under the old mentality, slot reservation fees, limited flexibility, while the clean rooms sit partially empty. The industry needs to come to grips with the fact that we're not in the booming days anymore. For most companies, the decision is outsourcing. You lose some control, but you gain reproducibility. If an independent CDMO can repeat your process, your value proposition goes up.
Justin Skoble: I agree. I can't imagine, in this economy, early-stage companies building their own manufacturing to support early-stage assets. The demand is too low and the math doesn't work. It becomes a matter of finding the right partner for your stage. Early-phase academic CDMOs can give you phase-appropriate capabilities and the attention and expertise you need to get first-in-human clinical data.
Moderator: Nikki, how do you integrate technology and automation early on?
Nikki Bishop: The easiest answer is: just start. A lot of what the industry does, it does because that's how it's always done. The commercial manufacturing bridge doesn't start at the end of the bridge; it starts in R&D. The misnomer is that commercial-ready technology is heavy and expensive and we can't afford it early. The reality is there are small things you can do while you're still figuring out if the science works. Simple digital data capture. Spreadsheets instead of paper. Capturing the decisions, not just the numbers; why did you make that choice? When you get to commercial manufacturing, you have the science nailed, and you're just making it scalable instead of transferring the science itself. Start simple, start small. Ask people who've been through commercial manufacturing what they wish they'd done earlier.
Gregg Nyberg: Building on that, picking a good partner early can help you understand what the journey looks like. One thing we do is engage with partners very early, often on a team supply run. They transfer the process, we run it, and we learn together what's going to be challenging to scale. You can think through bite-sized chunks: how do you run the process now in a way that sets up for automation and a streamlined commercial process later?
Moderator: What about the human side, capturing institutional knowledge from a lab or an academic setting and transferring it to another site?
Kumar Karyampudi: The mindset has to be that tech transfer is not a one-off activity; it's a collaboration between sending and receiving sites. Three things matter. First, tacit knowledge. Batch records and SOPs are standard, but institutional knowledge, how the site handles the product, what's worked, what hasn't, usually lives in people's heads. Document it. Second, you can't PowerPoint your way through a tech transfer. You have to have people shadowing. Receiving-site staff should spend time at the sending site. Sending-site staff should be there for the first two validation runs. Third, build redundancy. We've been manufacturing cellular products at Moffitt for 17 years. Some of my team have been there from the beginning, but I can't depend on one or two people being the experts. You need replicas of that expertise.
Justin Skoble: I'll echo that. We've done this very successfully, bringing folks from PD or MSAT at our CDMOs out to our facility, translating batch records into their language, having our people on-plant for every run, not just the first few. That builds the partnership and the camaraderie that makes the whole thing work.
Part 2: Phase 1 to Pivotal
Moderator: How do you balance phase appropriateness in your process and analytical development with the intent to go commercial eventually?
Yeh-Chuin Poh: Process and analytical development are absolutely critical. But you need to know what phase you're in. A lot of companies follow industry norms that, when you dig in, aren't actually regulatory requirements. Take potency assays. Critical for mechanism of action, critical for commercial, but technically, not required for your first-in-human filing. I've seen programs invest so heavily in potency assay development that they're years behind programs that prioritized showing clinical efficacy first. There's a balance. You want to be able to bridge to the future, but don't spend all your eggs now on things that cripple your timeline.
Kumar Karyampudi: When we support 15 INDs and have to think about which ones advance, it comes down to the signal from the early-phase trial. But you have to be careful: you need reasonable control on your CQAs and the parameters that drive them. Without that, you can't believe the data coming out of your trial, and you can't make a good decision about advancing.
Justin Skoble: One of the things that's difficult in advanced therapies; we think we have a product with CQAs that matter, but we don't really know until we've put them in patients and seen the responses. You learn from the translational data what "good" looks like. Until then, investing too early can give you more control over the wrong process.
Moderator: How do you balance speed with being genuinely ready to advance?
Gregg Nyberg: A lot of our clients need to get to the next value inflection point; it's existential. If you over-invest early, you may not have a company to get to the next stage. But you also don't want to cut corners and incur technology debt that forces expensive rework later. Quality-by-design helps you focus investment on the attributes most likely to matter. Don't try to optimize everything; focus on what has the most leverage.
Yeh-Chuin Poh: I think of it as three buckets: cost, quality, timeline. You can only optimize two. If you want cost and time, you compromise quality. If you want cost and quality, you compromise time. If you want time and quality, you pour money into it. Every program is tuning among those three throughout its life cycle.
Nikki Bishop: And the thing about flexibility: it really centers on standardization. What can you standardize? If you leave it too late, you don't have the flexibility you need because once you're commercial, you may want to manufacture in multiple sites in multiple ways. Find three things you can standardize on early: how you capture data, tools, templates, QbD standards; not to slow yourself down, but to help you go faster later.
Part 3: Preparing for Commercial
Moderator: From a CDMO perspective, what does a "good" tech transfer look like? And is it easier to transfer something at commercial stage?
Gregg Nyberg: An ideal client knows their product and process well. Realistically, phase-appropriate, younger companies don't know either perfectly yet. What matters most is that the client understands where their gaps are and is willing to engage. On commercial-stage transfers: it depends on how they got to that stage. If it was done in isolation from GMP realities, any change at commercial triggers significant comparability work. That's why partnering with your intended commercial CDMO early has real benefits.
Nikki Bishop: Nothing about tech transfer is easy at any stage. At commercial, you've proven the science; what you're transferring is confidence. You've proven it works. Can you transfer that confidence to another site while maintaining integrity? Technology is an enabler, but it's also the human interaction. Build those human relationships across your manufacturing partners.
Yeh-Chuin Poh: And there's regulatory. Changes in early phase are easier. At commercial, the bar is significantly higher. I've seen a commercial cell therapy still using manual hemocytometer counts, three operators, three hours, because switching to automated counting at commercial stage is such a high bar. Regulatory strategy should start at the beginning, not at commercial. At Semma Therapeutics, which Vertex acquired, our pluripotent stem cell program ran one continuous trial where Phase 1 efficacy data applied all the way through pivotal, because we didn't change anything material in between. If you change things, that same data doesn't travel.
Moderator: Who owns a tech transfer?
Kumar Karyampudi: Not one party. Both. Governance, accountability, and a defined path of escalation need to be aligned between both parties from the beginning. The sending site knows the process best; they have to transfer it accurately. The receiving site has to execute precisely and meet quality standards. Within each, you have manufacturing, analytics, QA; each owns part of the responsibility. If accountability isn't clear and governance isn't in place, the transfer drifts. Neither party owns success or failure alone.
Moderator: Final question: the FDA's recent move toward CMC flexibility for cell and gene therapy. Thoughts?
Kumar Karyampudi: Welcoming news. Early-stage hasn't really changed. At BLA stage, it looks like FDA is willing to work more flexibly with sponsors. That's good news.
Gregg Nyberg: The proof will be in the pudding. It was an announcement, not a guidance, and it's not clear yet that the day-to-day reviewers have absorbed it. A lot of what's in there is really leveraging tools that already exist, but with a commitment to double down on actually using them. It comes down to execution.
Q&A: Key Questions From the Panel
When should a program actually start planning its first tech transfer?
From day one of process development. The panelists were unanimous on this. If a startup intends to commercialize, and nearly all of them do, tech transfer to a CDMO is effectively inevitable, which means process and analytical development should be designed for portability from the earliest bench work. The caveat is phase appropriateness: you're building something transferable and GMP-compatible, not something commercial-grade. Early over-investment in commercial-grade processes is one of the most common ways young biotechs run out of runway before they have clinical data.
Build in-house manufacturing or outsource to a CDMO?
For the vast majority of early-stage programs in today's market, outsource. The economic calculus that justified in-house builds five or six years ago has shifted dramatically. Capacity is widely available, capital is more constrained, and the dose volumes required for dose-escalation studies don't support the cost of operating a dedicated facility. There's also a strategic benefit to outsourcing that's often missed: if an independent CDMO can reproduce your process, the value of your program, to investors, partners, and acquirers, goes up. In-house-only is a liability, not a moat.
How do you choose the right CDMO for your stage of development?
Match the partner to the program's current stage, not where you hope it'll be in three years. Early-phase academic and translational CDMOs often give first-in-human programs the attention, expertise, and phase-appropriate flexibility they need. You may not commercialize out of that facility, and you should plan for a future transfer, but the trade-off is usually the right one. As you move toward pivotal and commercial, the criteria shift: scalable GMP infrastructure, modality-specific experience, regulatory track record, and a partnership philosophy that treats tech transfer as a collaboration rather than a handoff become the filters that matter most.
How early should you start building digital and data infrastructure?
Earlier than most teams think, but simpler than most teams fear. You don't need an enterprise data fabric at the bench stage. What you need is a habit of capturing decisions alongside data: why a condition was changed, why a parameter was set, what the team learned in the last run. That knowledge is what makes future scale-up a translation problem instead of a re-discovery problem. Structured spreadsheets are a legitimate starting point. The sophistication grows in step with the program.
How do you capture tacit knowledge so it actually transfers?
Three practices, together. First, deliberately document tacit knowledge, what has worked, what hasn't, what your site has learned across prior programs. Second, invest in shadowing rather than slide decks: receiving-site staff spend time at the sending site, and sending-site staff are present on the floor for the first validation runs. Third, build redundancy. A single long-tenured expert is a vulnerability; institutional knowledge needs to live across multiple people to survive.
Do you need a fully qualified potency assay for Phase 1?
Technically no, it's not a regulatory requirement for first-in-human. The panel's shared view was that over-investing in analytical development before clinical proof-of-concept is one of the most common ways early programs burn resources and timelines. The balance: invest enough in your critical quality attribute framework that you can trust the signal you see in the clinic, but don't try to lock down everything before you have patients in trial. Comparability work can bridge later, as long as you're planning for it.
Is it easier to tech-transfer an early-stage or a near-commercial process?
It depends less on stage than on how the process was developed. A near-commercial process built in isolation from GMP realities can be harder to transfer than a deliberately phase-appropriate early-stage one, because any change at commercial stage triggers significant comparability work and regulatory scrutiny. The takeaway: partner early with the team you intend to commercialize with, so you're not re-engineering a process under time pressure at BLA stage.
Who owns the success of a tech transfer?
Both parties, and that's the point. No single group, sending or receiving, owns the outcome. What works is explicit governance, clear accountability across each sub-function (manufacturing, analytics, QA), and a defined path of escalation set from day one. Without those, the transfer drifts and issues turn into blame. With them, both sides know what success looks like and how to raise problems before they become failures.
How do you balance speed to clinic with being genuinely ready to advance?
Use a quality-by-design lens to focus investment where it has the most leverage. You can't optimize every attribute of every step; you'll run out of runway. What you can do is identify the handful of attributes and parameters most likely to be important, invest deeply there, and accept phase-appropriate trade-offs elsewhere. The thing to avoid is accumulating "technology debt," cut corners that force expensive rework at the next stage, when you have less time and more regulatory scrutiny.
What does the FDA's recent move toward CMC flexibility actually mean for programs?
Cautiously encouraging. The panel's consensus was that the announcement signals a real willingness to engage with sponsors more flexibly on CMC, particularly at BLA stage, but the practical change will depend on how day-to-day reviewers apply it. Much of what's in the announcement is a commitment to use tools that already exist, quality-by-design approaches, prior knowledge, platform data, more consistently. The proof, as one panelist put it, will be in the pudding.