AAV is one of the most widely used delivery platforms in gene therapy, supporting approved treatments for inherited retinal diseases, spinal muscular atrophy, hemophilia, and a growing number of clinical-stage programs.
While AAV manufacturing has become increasingly standardized, one critical bottleneck remains: the transfer construct carrying the gene of interest (GOI).
The Bottleneck: One Custom Component Holding Everything Up
Unlike helper and Rep/Cap plasmids, the GOI construct must be generated for every new program, often making it the longest lead-time component in the manufacturing workflow. That process takes 3-5 months for GMP material suitable for clinical use, placing it on the critical path for most AAV programs. Synthetic DNA offers an alternative approach, reducing timelines while potentially improving manufacturing performance.

Figure 1. The standard AAV production workflow has a GOI transfer plasmid bottleneck. While Helper and Rep/Cap plasmids are shelf-stable, recurring inputs, the GOI-encoding transfer plasmid often takes 3-5 months to obtain enough material to proceed to next steps.
What the Data Shows
In a head-to-head comparison of AAV8 production using identical helper and packaging inputs, the only variable changed was the source of the GOI construct. One process used a conventional plasmid transfer vector, while the other used an equivalent synthetic DNA construct.
The synthetic DNA construct produced an approximately 49% increase in viral genome titer relative to the plasmid control. Importantly, the full-to-empty capsid ratio remained comparable between conditions, indicating that the increase in yield was not achieved at the expense of vector quality.
For AAV developers, both metrics matter. Higher particle production alone has limited value if vector quality declines. Maintaining the full-to-empty ratio while increasing output suggests that the productivity gain is associated with productive vector generation rather than simply increased particle formation.
 Figure 2. AAV yield: oDNA vs. pDNA. oDNA delivered an ~49% higher viral genome titer than the matched plasmid control (statistically significant, p<0.01, n=2). Across the same comparison, the full-to-empty capsid ratio was maintained — meaning the yield increase did not come at the cost of packaging quality.
Figure 2. AAV yield: oDNA vs. pDNA. oDNA delivered an ~49% higher viral genome titer than the matched plasmid control (statistically significant, p<0.01, n=2). Across the same comparison, the full-to-empty capsid ratio was maintained — meaning the yield increase did not come at the cost of packaging quality.
Where the lift likely comes from. The proposed mechanism is upstream of vector assembly itself. Synthetic, enzymatically produced DNA provides a highly defined starting material, free of bacterial backbone sequences, antibiotic-resistance markers, endotoxin, and other plasmid-derived impurities. Unlike conventional plasmids, the platform also enables the incorporation of engineered adapter sequences that can be tailored to specific application requirements. While the contribution of each factor remains under investigation, the result was clear: higher vector yield without compromising full-to-empty quality.
Reading the Process: Landmark Bio’s Viral Vector Platform
To understand why the starting material matters, it helps to look at the process it feeds. AAV manufacturing follows a well-established workflow consisting of upstream vector production and downstream purification, formulation, and fill/finish operations. Landmark Bio’s viral vector capability is built on exactly this architecture and supports both AAV and LVV through a shared process and analytical backbone.
- Upstream. HEK293 producer cells are expanded and co-transfected with the GOI transfer construct, a Rep/Cap plasmid, and a helper plasmid to generate AAV particles.
- Downstream. Following harvest, the vector is purified through chromatography, concentrated and formulated by TFF, and prepared for sterile fill and finish.

Figure 3. AAV workflow: From first run to GMP. Upstream, HEK293 suspension cultures scale from small development vessels into 50L–200L bioreactors with growth and viability curves that track tightly across vessel sizes. Downstream runs clarification, capture and polishing chromatography, TFF-based concentration and formulation, and fully integrated sterile fill/finish. The titer panels, plotted at each downstream step, separate physical particles from functional vector — because the ratio between them is the real measure of process health.
.svg) Table 1. Landmark Bio analytics panel.
Table 1. Landmark Bio analytics panel.
Why analytics matter. A release vector is only as good as the analytics that characterize it. Two rows on the panel are especially relevant: residual plasmid and host-cell DNA. Anything carried in with the DNA input has to be measured out at the end. A cleaner, backbone-free input directly lightens the analytical and regulatory burden at release.
Why AAV + Synthetic DNA Is a Natural Fit
Not every manufacturing challenge has a clean solution. This one does. The fit between AAV production and synthetic DNA is structural, not incidental.
- The process is standardized. Synthetic DNA replaces the GOI transfer construct without changing the broader AAV workflow.
- The bottleneck is isolated. The GOI is often the only program-specific input.
- Design iteration becomes practical. Updated constructs can be generated in 3–4 weeks rather than months.
- Scale-up is no longer a capacity problem. Synthetic DNA scales without the constraints of bacterial fermentation.
- It is a platform that runs many products. Shorter GOI lead times can accelerate development across multiple programs.
Synthetic DNA in Two Weeks — and Everything That Comes with It
The Artis team (at Landmark Bio in Watertown, MA and Syngoi in Bilbao, Spain) produces optimized synthetic DNA (oDNA) through a cell-free enzymatic process, delivering pure, linear, double-stranded DNA encoding only the GOI — with no bacterial backbone, no antibiotic-resistance markers, and no endotoxin.³

Figure 4. End-to-end oDNA platform. Workflow schematic: customer starting material (existing pDNA or linear synthetic DNA) → restriction/adapter ligation → closed linear xDNA intermediate (repository) → rolling-circle amplification → finishing with custom adapters → gram-scale oDNA. Annotate the entire path as enzymatic, with no bacterial fermentation step. Gram-scale DNA delivery in as little as 2 weeks.
RUO and SP GMP-grade delivery: 3–4 weeks, compared to the 3–5 months typically required for traditional plasmid manufacturing.
Beyond speed, synthetic oDNA offers several advantages:
- Zero host-cell endotoxin. Cell-free production eliminates host-cell impurities.
- No antibiotic-resistance markers. Contains only the GOI and engineered adapters.
- No bacterial backbone. Only therapeutically relevant sequence.
- High batch-to-batch consistency. A defined synthetic process produces reproducible material.
- Engineered adapter sequences. Can be tailored to support specific design objectives.
From DNA to Vector — Under One Roof
Artis combines synthetic DNA production and vector manufacturing within the same organization, bringing together Syngoi's synthetic DNA capabilities and Landmark Bio's AAV and LVV manufacturing expertise.
Most programs manage DNA suppliers and CDMOs as separate relationships, creating additional handoffs, timelines, and points of failure. When DNA production and vector manufacturing are coordinated from the outset, development activities can run in parallel rather than in series. Key benefits include:
- Aligned timelines. DNA production and manufacturing schedules are coordinated from day one.
- Shared data and expertise. Technical questions are resolved within the same organization.
- Faster iteration. Construct updates can move directly into process development.
- Fewer handoffs. Reduced supply chain complexity and a single accountable partner.
- One quality framework. DNA production and vector manufacturing operate within an aligned quality system.
The result is a more streamlined path from GOI sequence to GMP vector production.

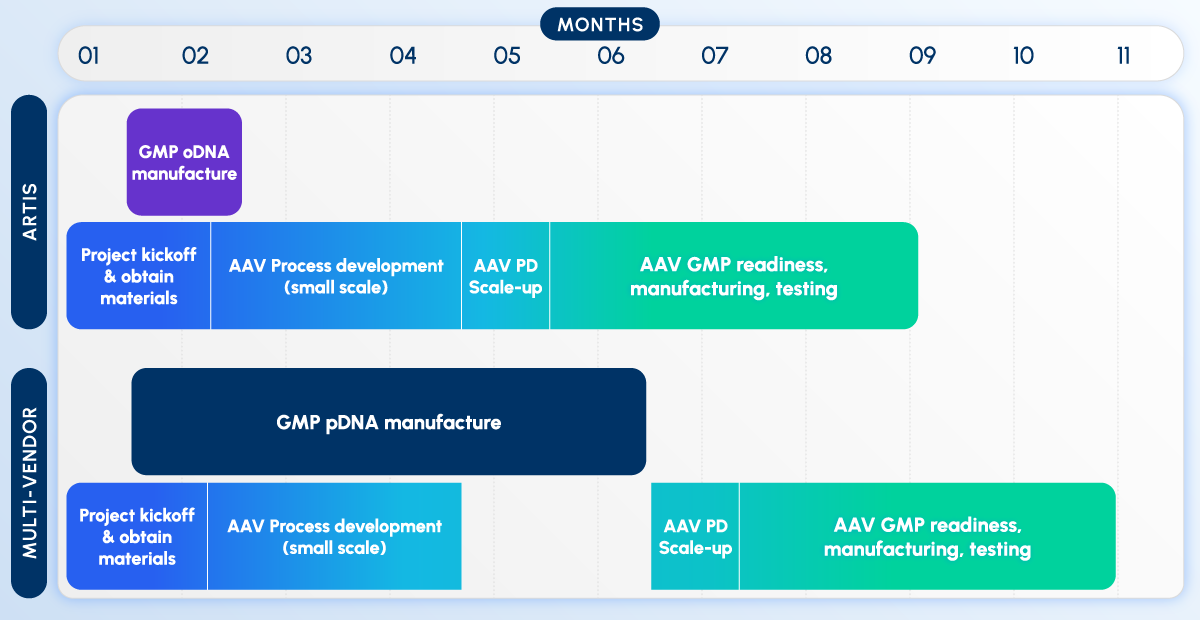
Figure 5. Same vector, two timelines. Top: Integrated synthetic DNA and manufacturing workflow. Bottom: Traditional supplier-to-CDMO workflow with sequential handoffs and longer lead times.
What Changes When Both Are Made In-House
The benefit of co-locating synthetic DNA and vector manufacturing is more than a shorter calendar. It changes the shape of the workflow itself.
Steps run in coordination instead of in series. In the conventional model, the program waits for plasmid release, ships material, re-qualifies on receipt, and only then books a manufacturing slot. When both sit in one organization, GOI synthesis and the manufacturing schedule are planned together from the outset.
One quality system spans both halves. A split supply chain means two quality systems, two release processes, and a re-incoming-inspection step at the boundary. Co-location lets DNA release and the vector campaign live under a single, aligned quality framework — fewer redundant tests, cleaner chain of custody.
A cleaner input simplifies downstream analytics. Because oDNA carries no bacterial backbone, no antibiotic-resistance gene, and no endotoxin, the residual-plasmid and host-related assays on the vector side start from a better baseline. That cleaner input also feeds into the full-to-empty story — the data above suggests yield can rise without compromising packaging quality.
Iteration loops close in weeks, not quarters. When process development flags a change — a tweak to the expression cassette, a codon-optimization adjustment, a different regulatory element — the same organization can synthesize the revised construct and feed it straight back into the next development run.
Fewer handoffs means less risk. Every transfer between organizations is a place where timelines slip, material is damaged in shipping, or accountability blurs when something goes wrong. Removing the supplier-to-CDMO boundary removes a whole category of failure modes.
Looking Ahead
Cell and gene therapy has moved from emerging technology to clinical reality, with a growing number of approved therapies and thousands of programs advancing through development. As demand for viral vectors continues to increase, pressure on development and manufacturing timelines is only expected to grow.
While vector manufacturing has advanced significantly, starting-material lead times remain a persistent challenge. By combining synthetic DNA and vector manufacturing within a single organization, Artis provides a faster, more integrated path from GOI to clinical-grade vector. In AAV production, this approach resulted in a 49% yield improvement while maintaining full-to-empty quality.
As development timelines accelerate, integrated DNA-to-vector workflows have the potential to reduce complexity, shorten timelines, and support the next generation of genetic medicines.
Ready to Accelerate Your AAV Program?
Whether you need synthetic DNA starting material, AAV manufacturing, or an integrated solution across both, Artis is ready to support your program from GOI to clinical-grade vector.
→ Explore Artis on Scientist.com
References
1. FDA, Approved Cellular and Gene Therapy Products. fda.gov
2. Cell & Gene Therapy Insights, scalable suspension HEK293 process development for viral vectors. insights.bio.
3. Artis BioSolutions to acquire Syngoi Technologies (Jan 6, 2026). businesswire.com.
4. U.S. Cell and Gene Therapy CDMO Market report (Nov 2025) — 2,000+ trials; market size and CAGR. globenewswire.com.
5. Viral Vectors & Plasmid DNA Manufacturing Market, SNS Insider (Dec 2025). globenewswire.com.
6. Grand View Research, Plasmid DNA Manufacturing Market — size, CAGR, segment shares. grandviewresearch.com.
7. Mordor Intelligence, Plasmid DNA Contract Manufacturing Market — downstream purification as bottleneck. mordorintelligence.com.